Stanovenie diagnózy idiopatická pľúcna fibróza. Diagnostika a liečba idiopatickej pľúcnej fibrózy
Zriedkavé choroby pľúca
Idiopatická pľúcna fibróza: moderný koncept a prístupy k diagnostike
S.N. Avdeev
Idiopatická pľúcna fibróza (IPF) je najviac časté ochorenie zo skupiny intersticiálnych pľúcnych ochorení a vyskytuje sa najmä u ľudí v strednom a staršom veku. Za posledné desaťročie došlo k významným zmenám nielen v prístupe k diagnostike IPF, ale aj v definícii IPF. Tento článok prezentuje moderné údaje o epidemiológii IPF, prezentuje výsledky nových štúdií patogenézy IPF, hlavné prístupy k diagnostike ochorenia, uvádza nové údaje o priebehu IPF a najčastejších sprievodných ochoreniach.
Kľúčové slová: intersticiálne pľúcne ochorenia, idiopatická pľúcna fibróza, klasifikácia, diagnostika, sprievodné ochorenia.
Idiopatická pľúcna fibróza (IPF) je najčastejším ochorením zo skupiny intersticiálnych pľúcnych ochorení (ILD) a vyskytuje sa najmä u ľudí v strednom a staršom veku. V priemere IPF predstavuje 20 až 30 % všetkých prípadov ILD, ochorenie má spravidla stabilne progresívny priebeh, čo vedie k rozvoju respiračné zlyhanie a smrť pacienta. Idiopatická pľúcna fibróza je jednou z foriem idiopatickej intersticiálnej pneumónie (IIP) a patrí medzi ne aj najčastejšie ochorenie.
Definícia IPF
Za posledné desaťročie došlo k významným zmenám nielen v prístupe k diagnostike IPF, ale aj v definícii IPF.
Je možné, že úplne prvý opis choroby, ktorá sa teraz nazýva IPF, predložil G.E. Rindfleisch v roku 1897 ako „cystická cirhóza pľúc“ (Cirrhosis cystica Pulmonum). Po mnoho rokov bola IPF (ďalšie synonymá pre IPF sú idiopatická fibrotizujúca alveolitída a kryptogénna fibrotizujúca alveolitída) definovaná ako progresívna fibrotická zápalové ochorenie pľúcny parenchým nejasného charakteru, ktorý zahŕňal množstvo podobných kli-
Ja Sergey Nikolaevich Avdeev - profesor, vedúci. Klinické oddelenie Federálnej štátnej rozpočtovej inštitúcie "Výskumný ústav pulmonológie" FMBA Ruska, Moskva.
niko-patologické stavy, v súčasnosti považované za samostatné ochorenia. Široká implementácia v klinickej praxi Počítačová tomografia s vysokým rozlíšením(HRCT) umožnila získať podrobnejší popis vzoru zmien v pľúcnom parenchýme a objasnila morfologickú klasifikáciu IIP, umožnila vykonať diferenciálnu diagnostiku medzi rôznymi ILD a viedla k viacerým presná definícia ILF.
Koncom 90. rokov niekoľko štúdií preukázalo vzťah medzi prognózou a rôznymi histopatologickými obrazcami IIP. Histopatologický obraz obvyklej intersticiálnej pneumónie (UIP) bol spojený s najslabšou prognózou v porovnaní s inými formami IIP, ako je nešpecifická intersticiálna pneumónia (NSIP) a deskvamatívna intersticiálna pneumónia (DIP). Obvyklá intersticiálna pneumónia je morfologický typ poškodenia pľúc s pestrým obrazcom, pri ktorom sa v pľúcnom parenchýme striedajú oblasti normálneho a patologicky zmeneného pľúcneho tkaniva, t.j. dočasná heterogenita fibrózy pozostávajúca z ložísk fibroblastických ložísk umiestnených medzi acelulárnym hustým fibróznym tkanivom, čo vedie k rozvoju deformácie architektoniky parenchýmu a vzniku „plástových pľúc“ (obr. 1).
Tieto zmeny prevládajú v subpleurálnych a paraseptálnych zónach pľúc. Fib-
Roblastické ložiská sa zvyčajne nachádzajú na hranici medzi oblasťami s fibrotickými zmenami a normálnym pľúcnym parenchýmom.
V roku 2000 medzinárodný konsenzuálny dokument definoval IPF ako histopatologický vzorec UIP neznámeho pôvodu, t.j. pri absencii známej príčiny poškodenia pľúc ako technika lieky, vystavenie inhalačným a pracovným faktorom, radiačnej terapii a systémovým ochoreniam spojivové tkanivo. Táto definícia sa používa dodnes.
Epidemiológia
V Spojených štátoch sa incidencia IPF pohybuje od 7 do 17 na 100 000 ľudí za rok, zatiaľ čo prevalencia IPF sa pohybuje od 20 do 60 ľudí na 100 000 vo všeobecnej populácii. Priemerný vek pacientov v čase diagnózy IPF sa pohybuje od 50 do 85 rokov. Intersticiálna pľúcna fibróza je pomerne zriedkavá u pacientov mladších ako 50 rokov (celkový podiel medzi pacientmi s IPF sa pohybuje od 2 do 15 %). Pacienti s IPF sú prevažne muži s pomerom mužov a žien približne 1,5:1. 1 – 4 % všetkých pacientov s IPF má v rodinnej anamnéze pľúcnu fibrózu. V porovnaní so sporadickými formami IPF sa u mladších pacientov vyskytujú familiárne formy pľúcnej fibrózy.
Podľa epidemiologických štúdií sa ukazuje súvislosť IPF s fajčením, ako aj s expozíciou organickým a anorganickým druhom prachu, farmakologickou terapiou, infekčnými faktormi, ako napr. vírus Epstein-Barr. Napriek tomu veľké číslo Hoci štúdie preukázali tieto asociácie, úloha týchto látok v etiológii IPF zostáva nejasná.
Patogenéza
Príčiny IPF stále zostávajú neznáme. Neúčinnosť protizápalovej liečby v liečbe IPF, vrátane vysokých dávok glukokortikosteroidov (GCS), viedla k pochybnostiam o vedúcej úlohe chronický zápal pri vzniku parenchýmovej fibrózy pri tomto ochorení. V súčasnosti sa všeobecne uznáva, že hlavným mechanizmom vedúcim k rozvoju progresívnej pľúcnej fibrózy je opakované a pretrvávajúce poškodenie. alveolárny epitel s ich následnou dysregulovanou obnovou. Hlavné bunky
Ryža. 1. Idiopatická pľúcna fibróza: histologický obraz bežnej intersticiálnej pneumónie (šípka).
Bunky zodpovedné za rozvoj fibrotických zmien v pľúcach sú myofibroblasty a ich prekurzory. Mechanizmy, ktoré sú základom náboru a proliferácie týchto buniek, je potrebné objasniť, ale už je známe, že sú sprostredkované veľká kvantita mediátory, vrátane cytokínov, chemokínov, fibrogénnych faktorov, koagulačných proteínov, oxidantov a regulátorov apoptózy. Je pravdepodobné, že integrálnym článkom vo fibrotickom procese je ukladanie zložiek extracelulárnej matrice. Vzhľadom na to, že IPF typicky postihuje ľudí v strednom a staršom veku, možno predpokladať, že biologické zmeny súvisiace s vekom, ako napríklad zmeny funkcie telomér, zohrávajú úlohu aj pri vzniku IPF.
Tieto procesy môžu viesť k predčasnému bunkovému starnutiu alveolárnych buniek a vyčerpaniu progenitorových buniek potrebných na alveolárnu regeneráciu, čo vedie k aberantnej oprave prostredníctvom rozvoja fibrózy. Aj dnes sa zvažujú hypotézy, podľa ktorých hrá hlavnú úlohu pri vzniku pľúcnej fibrózy mechanické namáhanie, napríklad opakované pôsobenie ťažných síl na perifériu „starnúcich“ pľúc.
Určitý pokrok sa dosiahol v identifikácii genetických determinantov pľúcnej fibrózy. Napríklad nedávne genómové štúdie identifikovali asociáciu IPF s jednonukleotidovým alelickým variantom promótora génu MiC5B, ktorý je prítomný u 38 % pacientov s IPF. Je zaujímavé, že prítomnosť alelického variantu promótora génu MiC5B bola spojená s lepšou prognózou u pacientov s IPF. Okrem toho množstvo štúdií ukázalo, že varianty v génoch pre telomerázové zložky boli spojené s vývojom
|Obr. 2. Idiopatická pľúcna fibróza: CT obraz intersticiálnej pneumónie (retikulárne zmeny, trakčné bronchiektázie a „medové“ zmeny).
máme radi rodinné formy pľúcna fibróza a IPF. Ďalšie gény zapojené do vývoja familiárnej pľúcnej fibrózy zahŕňajú gény surfaktantového proteínu C a A2.
Klinický obraz
Hlavnými sťažnosťami väčšiny pacientov s IPF sú progresívna dýchavičnosť a suchý kašeľ. Menej časté príznaky sú nepríjemné pocity na hrudníku a takzvané konštitučné príznaky (únava, horúčka nízkeho stupňa a chudnutie). U niektorých pacientov s IPF sú prvé nálezy respiračné symptómy, ale zmeny v pľúcnych funkčných parametroch alebo údajoch HRCT.
Takmer u všetkých pacientov s IPF je pri auskultácii počuť inspiračný krepitus v posterobazálnych častiach pľúc, ktorý je opísaný ako „Velero rales“; približne polovica všetkých pacientov má zmeny na terminálnych falangách prstov vo forme „ paličky“. U pacientov s pokročilými zmenami, fyzickými známkami pľúcnej hypertenzie (PH) a pľúcne srdce, ako je akcent druhého tónu nad pľúcnou tepnou, systolický šelest trikuspidálnej regurgitácie, periférny edém. Cyanóza a periférny edém sú tiež neskorými príznakmi IPF.
Laboratórne testy
Všeobecná analýza krv môže vykazovať zrýchlené ESR, spravidla však hladina he-
Moglobín a celkový počet krvných leukocytov sú v rámci normálnych hodnôt. Niektorí pacienti s IPF majú zvýšené titre antinukleárnych protilátok, reumatoidného faktora alebo iných autoprotilátok, ale zároveň nie je možné zistiť prítomnosť systémové ochorenia spojivového tkaniva (CTT).
V posledných rokoch boli identifikované potenciálne diagnostické a prognostické biomarkery periférnej krvi špecifické pre IPF. Tieto biomarkery zahŕňajú metaloproteinázy MMP-1 a MMP-7, chemokín CCL-18 (Chemokine (C-C motív) ligand 18), surfaktantový proteín A, proteín podobný chitináze YKL-40, voľnú cirkulujúcu deoxyribonukleovú kyselinu, periostín a osteopontín. Krebs von den Lungen faktor 6 (KL-6) je mucínový glykoproteín s vysokou molekulovou hmotnosťou, ktorý je exprimovaný hlavne na pneumocytoch typu II a epitelových bunkách respiračných bronchiolov. Hladiny KL-6 v sére sú markerom poškodenia alveolárnych epitelových buniek a možno ich použiť na posúdenie závažnosti ILD, vrátane IPF. Marker alveolomucín (3EG5) má podobnú štruktúru ako KL-6.
Röntgenová snímka IPF
Rádiografia hrudník u pacientov s IPF najčastejšie odhalí bilaterálne retikulárne zmeny (zvýšený pľúcny vzor) v dolných a periférnych zónach pľúc. Až 10 % pacientov s IPF má však úplne normálny rádiografický nález. S progresiou ochorenia sa retikulárne zmeny stávajú hrubšími, pľúcne objemy sa zmenšujú av niektorých prípadoch sa rozlišujú periférne zmeny typu „voštinových pľúc“ a príznaky PH, ako je rozšírenie priemeru pľúcnej artérie a kardiomegália.
Jednou z hlavných metód potrebných na diagnostiku IPF je HRCT. Táto metóda umožňuje získať detailný obraz o zmenách vnútrohrudných štruktúr a je často dostatočnou diagnostickou metódou na potvrdenie niektorých foriem ILD. CT vyšetrenie vysoké rozlíšenie je citlivejšie ako röntgen hrudníka a je vhodnejšie pre odlišná diagnóza rôzne formy IZL.
Spoľahlivá rádiologická diagnóza AIP môže byť stanovená v prítomnosti dvoch
retikulárne opacity tretích strán v kombinácii s trakčnou bronchiektáziou/bronchioloektáziou, hlavne v subpleurálnych oblastiach a v prítomnosti subpleurálnych „voštinových“ zmien (obr. 2). Je však potrebné poznamenať, že v niektorých prípadoch môže byť „medové“ zmeny veľmi ťažké odlíšiť od trakčnej bronchiolektázy, subpleurálnych cýst a paraseptálneho emfyzému. Ak sú prítomné zmeny zábrusu, ich rozsah by mal byť menší ako rozsah retikulárnych zmien. Tiež pri IPF môže dôjsť k miernemu zvýšeniu veľkosti lymfatické uzliny, ale zvyčajne nie sú žiadne zmeny v pohrudnici. Ak sú tieto charakteristiky prítomné, rádiologická diagnóza AIP vo viac ako 90 % prípadov zodpovedá morfologickej.
Testy funkcie pľúc
Pľúcne funkčné testy u pacientov s IPF zvyčajne odhalia reštriktívne zmeny v pľúcach s poklesom pľúcne objemy a zníženie difúznej kapacity pľúc. Zapnuté skoré štádia IPF môže vykazovať izolovaný pokles pľúcnej difúznej kapacity v prítomnosti normálnych pľúcnych objemov. Jeden z skoré príznaky narušenie výmeny plynov je expanzia alveolo-arteriálneho kyslíkového gradientu. Aj s normálne hladiny saturácia krvi kyslíkom za pokojových podmienok, fyzická aktivita vedie k desaturácii, t.j. znížené hladiny okysličovania, čo je tiež charakteristické pre IPF.
Pri kombinácii IPF a emfyzému dochádza k relatívnej normalizácii pľúcnych objemov a prietokov. Teda u pacienta s ťažkou dýchavičnosťou počas fyzická aktivita spirografia a telesná pletyzmografia vykazujú prakticky nezmenené funkčné ukazovatele. V takýchto situáciách sa spravidla zistí významný pokles difúznej kapacity pľúc a HRCT umožňuje zistiť v rovnakom pľúca pacienta pľúcna fibróza (v bazálnych oblastiach) a emfyzém (in horné časti) .
Kritériá diagnózy pre IPF
Definitívna diagnóza IPF vyžaduje dôkaz o vzore AIP, buď pomocou HRCT alebo chirurgickej pľúcnej biopsie (ak existujú zmeny HRCT, ktoré nie sú konzistentné s AIP, ako je horná prevaha, konsolidácia mozaiky, difúzne mikronoduly) a
1. Vylúčte známe príčiny ILD (napr. expozícia fibrogénnym látkam z povolania alebo prostredia, CTD, liekom a liečenie ožiarením)
2. Vzor IIP podľa:
Chirurgická pľúcna biopsia v prítomnosti zmien podľa HRCT údajov, ktoré nie sú charakteristické pre AIP
s vylúčením známych príčin ochorenia, ako sú inhalačné faktory (napríklad chronická exogénna alergická alveolitída (EAA) a azbestóza) a CTD (tabuľka).
Počítačová tomografia s vysokým rozlíšením dokáže presne diagnostikovať AIP približne u dvoch tretín pacientov s IPF. U pacientov s atypickými HRCT nálezmi je potrebné ďalšie vyšetrenie na potvrdenie IPF alebo iných foriem ILD. Hoci sa transbronchiálna biopsia (TBB) vo všeobecnosti považuje za nevhodnú metódu na morfologickú verifikáciu AIP, TBB a bronchoalveolárna laváž (BAL) môžu potvrdiť ďalšie ochorenia zo skupiny ILD, ako sú sarkoidóza, EAA, eozinofilná pneumónia, histiocytóza z Langer-Hansových buniek a pľúcna alveolárna proteinóza Napríklad tekutinová lymfocytóza BAL (>30 % lymfocytov) u pacienta s podozrením na IPF naznačuje alternatívnu diagnózu, ako je nešpecifická intersticiálna pneumónia alebo EAA. S príchodom novej metódy odberu pľúcneho materiálu – transbronchiálnej kryobiopsie, ktorá umožňuje získať väčší objem kúskov pľúcneho tkaniva s menšou deformáciou v porovnaní s použitím tradičných bioptických klieští, existuje nádej, že metódy bronchoskopickej biopsie bude hrať významnejšiu úlohu diagnostický algoritmus ILF.
V prípade potreby sa na potvrdenie diagnózy IPF vykoná chirurgická pľúcna biopsia buď video-asistovanou torakoskopiou alebo torakotómiou. Na získanie reprezentatívnych vzoriek pľúcneho tkaniva sa vykonávajú chirurgické pľúcne biopsie z rôznych lalokov pľúc. Hoci chirurgická pľúcna biopsia sa považuje za najviac presná metóda stanovenie histopatologického obrazu ILD, samotný výkon je spojený s určitým rizikom nežiaducich účinkov, najmä u pacientov s ťažkým funkčné zmeny, rýchle zhoršenie stavu a prítomnosť sprievodných
Ryža. 3. Exacerbácia IPF: CT vyšetrenie pľúc 2 mesiace pred exacerbáciou (a) a počas exacerbácie IPF (b).
ďalšia patológia. Rozhodnutie podstúpiť chirurgickú pľúcnu biopsiu sa preto musí urobiť individuálne, berúc do úvahy klinický obraz, diagnostické možnosti, potenciálne prínosy dosiahnutia definitívnej diagnózy, zváženie rizika postupu a preferencie pacienta.
Keďže diagnóza IPF vyžaduje klinické, rádiologické a morfologické údaje, multidisciplinárna zhoda týchto nálezov zvyšuje diagnostickú presnosť. Nedávny medzinárodný konsenzus o diagnostike a manažmente IPF zdôrazňuje, že okrem získania kritérií pre AIP z HRCT a pľúcnej biopsie, presná diagnóza IPF potrebuje stanovisko multidisciplinárnej rady. Akékoľvek HRCT nálezy, ktoré sú atypické pre IPF, by mali zvýšiť možnosť diagnózy IPF, aj keď chirurgická pľúcna biopsia odhalila morfologický vzorec AIP. Napríklad choroby, ako je chronická EAA, pľúcne lézie vyvolané liekmi, CTD a azbestóza, môžu mať podľa chirurgickej pľúcnej biopsie morfologický vzor AIP.
Klinický priebeh a komorbidné stavy
Priemerná miera prežitia pacientov s IPF je približne 3 roky. Napriek tomu, že u mnohých pacientov zvyčajne dochádza k postupnej, stabilnej progresii ochorenia v podobe narastajúcej dýchavičnosti a zhoršenia funkčných pľúcnych parametrov, je takmer nemožné individuálne predpovedať priebeh IPF. Niektorí pacienti nezaznamenajú žiadne zhoršenie klinických a funkčných parametrov počas mesiacov alebo rokov, zatiaľ čo u iných pacientov môže dôjsť k náhlemu, rýchlemu zhoršeniu s rozvojom progresívneho respiračného zlyhania. Okrem toho boli pri IPF opísané rôzne vzorce progresie ochorenia, „pomalá“ a „rýchla“, a podľa pilotnej štúdie existujú určité genetické determinanty rýchlej progresie IPF.
Na zlepšenie predpovede prognózy pacientov s IPF bolo navrhnutých niekoľko systémov na určovanie závažnosti alebo štádia IPF, ale žiadny zatiaľ nezískal rozsiahle klinické schválenie. Progresívne respiračné zlyhanie je príčinou úmrtia približne polovice pacientov s IPF, zatiaľ čo medzi ďalšie príčiny patrí pneumónia, aspirácia, infarkt myokardu, cievna mozgová príhoda a iné mimopľúcne príčiny.
Exacerbácie IPF
U pacientov s IPF s relatívne pomalou progresiou ochorenia možno pozorovať akútne epizódy zhoršenia klinického obrazu s rozvojom ťažkého respiračného zlyhania, ktoré často vedie k smrti pacienta. Niektoré z týchto epizód sú exacerbácie IPF, ktoré sú definované ako rýchle zhoršenie dýchavičnosti (v priebehu posledných 30 dní: zhoršenie výmeny plynov a nové zmeny na RTG/HRCT pri absencii identifikovanej príčiny zhoršenia, ako napr. infekcia, srdcové zlyhanie alebo tromboembolizmus pľúcne tepny). Zmeny HRCT spojené s exacerbáciou IPF zahŕňajú „nové“ zákalové opacity superponované na zmeny charakteristické pre pľúcnu fibrózu (voštinové a retikulárne zmeny) (obrázok 3).
Fenomén exacerbácií IPF ešte nie je dostatočne preskúmaný, môžu sa vyskytnúť kedykoľvek v priebehu ochorenia a niekedy sú prvým prejavom IPF. Vo všeobecnosti pacienti s klinický obraz exacerbácia
Pri IPF sa odporúča BAL na vylúčenie infekcií. Chirurgická pľúcna biopsia odhaľuje obraz difúzneho alveolárneho poškodenia superponovaného na obrazec AIP, ale pľúcna biopsia sa zvyčajne neodporúča, keď sa rozvinie exacerbácia IPF. Úmrtnosť pacientov s rozvojom exacerbácie IPF je napriek užívaniu kortikosteroidov a antibiotík stále veľmi vysoká – 70 – 80 %.
Kombinácia pľúcnej fibrózy a emfyzému
Niektorí pacienti majú okrem IPF aj ďalšie ochorenie pľúc – emfyzém, táto kombinácia sa nazýva kombinácia pľúcnej fibrózy a emfyzému (CPFE). Prebieha diskusia o tom, či sú pacienti jedinečne predisponovaní k rozvoju fibrózy aj emfyzému, alebo či je náhoda, že sa pľúcna fibróza vyvinie u fajčiara s už existujúcim emfyzémom. Pacienti s CLFE sú zvyčajne muži s významnou anamnézou fajčenia. U takýchto pacientov môže HRCT odhaliť emfyzém v horných častiach pľúc a UIP v dolných častiach. Niektoré štúdie ukázali, že CLFE je spojený s vysokým výskytom PH a horšou prognózou, dokonca aj v porovnaní s IPF.
Pľúcna hypertenzia je relatívne častou komplikáciou IPF sa nachádza celkovo približne u 30 – 40 % pacientov au 85 % pacientov s terminálne štádiá choroby.
Pľúcna hypertenzia u pacientov s IPF je zvyčajne spojená s ťažkou pľúcnou dysfunkciou a hypoxémiou, ale niekedy sa vyskytuje aj pri ľahších formách ochorenia. Analýza nedávnych prípadov vyvoláva otázku možnosti rozvoja prestavby pľúcne cievy a bez účasti hypoxickej vazokonstrikcie, ale v dôsledku fibrotickej reštrukturalizácie pľúcneho parenchýmu.
Pľúcna hypertenzia u pacientov s CLPE môže byť spojená so zmenšením celkovej plochy kapilár v dôsledku fibrotickej a emfyzematóznej deštrukcie pľúcneho cievneho riečiska. K rozvoju PH u niektorých pacientov s IPF prispieva aj desaturácia počas spánku.
Prítomnosť PH u pacientov s IPF vedie k ďalšiemu poklesu fyzickej výkonnosti a je spojená so zlou prognózou
Pacientom s PH a hypoxémiou sa zvyčajne predpisuje oxygenoterapia; Stále neexistujú žiadne formálne dôkazy, že takáto liečba zlepšuje prežívanie pacientov s IPF. V nedávno uskutočnenom klinická štúdia Nepreukázalo sa, že liečba sildenafilom zvyšuje vzdialenosť v 6-minútovom teste chôdze (6-MT) (primárny koncový bod) u pacientov s ťažkou IPF (difúzna kapacita<35% от должной), однако данная терапия привела к улучшению параметров газообмена, уменьшению одышки и улучшению качества жизни . По данным проведенных исследований, ни один из препаратов группы антагонистов рецепторов эндотелина (бозентан, амбризентан, мацитентан) не оказывал положительного влияния на функциональные показатели, прогрессирование заболевания или выживаемость больных .
Gastroezofageálna refluxná choroba
Gastroezofageálny reflux (GERD) sa vyskytuje u veľkej väčšiny pacientov s IPF (prevalencia sa pohybuje od 67 do 94 %), čo je oveľa častejšie ako u pacientov s inými pľúcnymi ochoreniami, ako je astma, chronická obštrukčná choroba pľúc alebo iné ILD. . V posledných rokoch sa široko diskutuje o vzťahu medzi GERD a IPF: je možné, že mikroaspirácia kyslého obsahu žalúdka môže viesť k poškodeniu alveolárneho epitelu, t.j. počiatočná väzba v patogenéze IPF. U pacientov s exacerbáciou IPF sa v tekutine BAL nachádzajú zvýšené koncentrácie pepsínu, čo naznačuje, že primárnym spúšťačom tejto komplikácie je aspirácia.
Nedávna retrospektívna štúdia ukázala, že antirefluxná liečba u pacientov s IPF je spojená s pomalšou progresiou pľúcnych funkčných testov a lepším prežívaním pacientov. U pacientov s IPF s príznakmi GERD čakajúcich na transplantáciu pľúc stabilizovala fundoplikácia Nissen požiadavky na kyslík v porovnaní s pacientmi, ktorí nepodstúpili operáciu, hoci parametre funkcie pľúc zostali medzi týmito skupinami pacientov porovnateľné. A u pacientov, ktorí podstúpili transplantáciu pľúc, fundoplikácia umožnila spomaliť progresiu bronchiolitis obliterans v r.
prípad dokumentovaného mechanického refluxu alebo symptomatickej GERD.
Rakovina pľúc
Riziko vzniku rakoviny pľúc je výrazne zvýšené u pacientov s IPF, najmä u starších ľudí s dlhou anamnézou fajčenia. U pacientov s IPF je pomerne ťažké odhaliť karcinóm pľúc pre fibrotické zmeny v pľúcnom tkanive, typickým obrazom nádoru sú nodulárne zmeny s nerovnomernou alebo ihličkovitou kontúrou v periférnych zónach pľúc. Diagnózu rakoviny pľúc možno stanoviť buď pred diagnózou IPF, alebo po diagnóze IPF alebo dokonca súčasne s ňou.
Funkčné poškodenie v dôsledku IPF predstavuje riziko pre chirurgickú liečbu rakoviny pľúc, pretože prítomnosť IPF je spojená s vyššou pooperačnou morbiditou a mortalitou u pacientov v porovnaní s pacientmi bez IPF. Okrem toho je známe, že chirurgické zákroky na pľúcach u pacientov s IPF môžu viesť k rozvoju exacerbácie IPF, ktorá je spojená s vysokou mortalitou pacientov.
Venózna tromboembólia
Populačné štúdie naznačujú, že pacienti s IPF majú zvýšené riziko vzniku venózneho tromboembolizmu (VTE). Predisponujúcimi faktormi k VTE pri IPF môže byť nielen obmedzená aktivita pacienta, ale aj zvýšená prokoagulačná aktivita, ktorá tiež prispieva k procesu fibroformácie. Okrem toho je potrebné poznamenať, že úmrtia u pacientov s IPF a VTE nastávajú v skoršom veku ako u pacientov s IPF bez VTE. Venózna tromboembólia je jednou z príčin exacerbácie respiračného zlyhania u pacientov s IPF. V jednej retrospektívnej štúdii pacientov s IPF prijatých na jednotku intenzívnej starostlivosti s respiračným zlyhaním bola pľúcna embólia hlavnou príčinou zhoršenia v 6 % prípadov.
Bibliografia
1. Raghu G. a kol. //Dopom. J. Respira. Crit. Care Med. 2011.
2. American Thoracic Society // Am. J. Respira. Crit. Care Med.
2000. V. 161. S. 646.
3. Ryu J.H. a kol. // Mayo Clin. Proc. 2014. V. 89. S. 1130.
4. Travis W.D. a kol. //Dopom. J. Respira. Crit. Care Med. 2013.
5. Collard H.R. a kol. //Arch. Stážista. Med. 2003. V. 163. S. 17.
6. American Thoracic Society // Am. J. Respira. Crit. Care Med. 2002. V. 165. S. 277.
7. Katzenstein A.L. a kol. //Dopom. J. Respira. Crit. Care Med. 1998. V. 157. S. 1301.
8. Rindfleisch G.E. // Centralbl. Pathol. 1897. V. 8. S. 864.
9. Ilkovič M.M. a ďalšie // Pulmonológia. 2003. Číslo 3. S. 98.
10. Ilkovič M.M. a iné // Consilium medicum. 2009. Číslo 11. S. 24.
11. Bjoraker J.A. a kol. //Dopom. J. Respira. Crit. Care Med. 1998. V. 157. S. 199.
12. Nagai S. a kol. //Eur. Respira. J. 1998. V. 12. S. 1010.
13. Daniil Z.D. a kol. //Dopom. J. Respira. Crit. Care Med. 1999. V. 160. S. 899.
14. Selman M. a kol. //Ann. Stážista. Med. 2001. V. 134. S. 136.
15. du Bois R.M. //Nat. Rev. Drug. Discov. 2010. V. 9. S. 129.
16. Fernández Pérez E.R. a kol. // Hrudník. 2010. V. 137. S. 129.
17. Raghu G. a kol. //Dopom. J. Respira. Crit. Care Med. 2006. V. 174. S. 810.
18. Douglas W.W. a kol. //Dopom. J. Respira. Crit. Care Med. 2000. V. 161. S. 1172.
19. Kráľ T.E. a kol. //Dopom. J. Respira. Crit. Care Med. 2001. V. 164. S. 1171.
20. Nadrous H.F. a kol. // Mayo Clin. Proc. 2005. V. 80. S. 37.
21. Allam J.S. a kol. //Curr. Opin. Pulm. Med. 2006. V. 12. S. 312.
22. Steele M.P. a kol. //Dopom. J. Respira. Crit. Care Med. 2005. V. 172. S. 1146.
23. Lee H.-L. a kol. // Hrudník. 2005. V. 127. S. 2034.
24. Wahidi M.M. a kol. // Hrudník. 2002. V. 121. S. 30S.
25. Taskar V. a kol. // Semin. Respira. Crit. Care Med. 2008. V. 29. S. 670.
26. Blackwell T.S. a kol. //Dopom. J. Respira. Crit. Care Med. 2014. V. 189. S. 214.
27. Rosas I.O. a kol. //Dopom. J. Respira. Crit. Care Med. 2013. V. 188. S. 765.
28. Phan S.H. //Proc. Am. Thorac. Soc. 2012. V. 9. S. 148.
29. Xia H. a kol. //Dopom. J. Pathol. 2014. V. 184. S. 1369.
30. Noble P.W. a kol. // J. Clin. investovať. 2012. V. 122. S. 2756.
31. Maher T.M. //Curr. Opin. Pulm. Med. 2013. V. 19. S. 460.
32. Shimbori C. a kol. //Curr. Opin. Pulm. Med. 2013. V. 19. S.446.
33. Jelša J.K. a kol. //Proc. Natl. Akad. Sci. USA. 2008. V. 105. S. 13051.
34. Chilosi M. a kol. //Prekl. Res. 2013. V. 162. S. 156.
35. Leslie K.O. //Arch. Pathol. Lab. Med. 2012. V. 136. S. 591.
36. Seibold M.A. a kol. // N. Engl. J. Med. 2011. V. 364. S. 1503.
37. Peljto A.L. a kol. // JAMA. 2013. V. 309. S. 2232.
38. Armanios M.Y. a kol. // N. Engl. J. Med. 2007. V. 356. S. 1317.
39. Cronkhite J.T. a kol. //Dopom. J. Respira. Crit. Care Med. 2008. V. 178. S. 729.
40. Liu T. a kol. //Dopom. J. Respira. Cell Mol. Biol. 2013. V. 49. S. 260.
41. Thomas A.Q. a kol. //Dopom. J. Respira. Crit. Care Med. 2002. V. 165. S. 1322.
42. Wang Y. a kol. //Dopom. J.Hum. Genet. 2009. V. 84. S. 52.
43. Behr J. a kol. //Eur. Respira. J. 2008. V. 31. S. 1357.
44. Nathan S.D. a kol. //Dopom. J. Respira. Crit. Care Med. 2007. V. 175. S. 875.
45. Zhang Y. a kol. //Curr. Opin. Pulm. Med. 2012. V. 18. S. 441.
46. Vij R. a kol. //Prekl. Res. 2012. V. 159. S. 218.
47. Tzouvelekis A. a kol. // Respira. Res. 2005. V. 6. S. 78.
48. Prasse A. a kol. // Respirológia. 2009. V. 14. S. 788.
49. Avdeeva O.E. a ďalšie // Pulmonológia. 1998. Číslo 2. S. 22.
50. Silva C.I. a kol. // J. Thorac. Zobrazovanie. 2009. V. 24. S. 260.
51. Lynch D.A. a kol. //Dopom. J. Respira. Crit. Care Med. 2005. V. 172. S. 488.
52. Arakawa H. a kol. // A.J.R. Am. J. Roentgenol. 2011. V. 196. S. 773.
54. Cottin V. a kol. //Eur. Respira. J. 2005. V. 26. S. 586.
56. Ryerson C. J. a kol. // Hrudník. 2013. V. 144. S. 234.
57. Trahan S. a kol. // Hrudník. 2008. V. 134. S. 126.
58. Misumi S. a kol. //Proc. Am. Thorac. Soc. 2006. V. 3. S. 307.
59. Ryu J.H. a kol. // Mayo Clin. Proc. 2007. V. 82. S. 976.
60. Leslie K.O. a kol. //Arch. Pathol. Lab. Med. 2007. V. 131. S. 407.
61. Ohshimo S. a kol. //Dopom. J. Respira. Crit. Care Med. 2009. V. 179. S. 1043.
62. Casoni G.L. a kol. // PLoS One. 2014. V. 9. P. e86716.
63. Maldonado F. a kol. // J. Bronchology Interv. Pulmonol. 2009. V. 16. S. 227.
64. Riley D.J. a kol. //Curr. Opin. Pulm. Med. 2006. V. 12. S. 331.
65. Sharma S. // Curr. Opin. Pulm. Med. 2012. V. 18. S. 528.
66. Utz J.P. a kol. //Eur. Respira. J. 2001. V. 17. S. 175.
67. Park J.H. a kol. //Eur. J. Cardiothorac. Surg. 2007. V. 31. S. 1115.
68. Flaherty K.R. a kol. //Dopom. J. Respira. Crit. Care Med. 2004. V. 170. S. 904.
69. Smith M. a kol. // J. Clin. Pathol. 2013. V. 66. S. 896.
70. Ley B. a kol. //Dopom. J. Respira. Crit. Care Med. 2011. V. 183. S. 431.
71. Selman M. a kol. // PLoS One. 2007. V. 2. P. e482.
72. Wells A.U. a kol. //Dopom. J. Respira. Crit. Care Med. 2003. V. 167. S. 962.
73. Ley B. a kol. //Ann. Stážista. Med. 2012. V. 156. S. 684.
74. du Bois R.M. a kol. //Dopom. J. Respira. Crit. Care Med. 2011. V. 184. S. 459.
75. Daniels C.E. a kol. //Eur. Respira. J. 2008. V. 32. S. 170.
76. Panos R.J. a kol. //Dopom. J. Med. 1990. V. 88. S. 396.
77. Collard H.R. a kol. //Dopom. J. Respira. Crit. Care Med. 2007. V. 176. S. 636.
78. Parambil J.G. a kol. // Hrudník. 2005. V. 128. S. 3310.
79. Saydain G. a kol. //Dopom. J. Respira. Crit. Care Med. 2002. S. 166. S. 839.
80. Kim D.S. // Clin. Chest Med. 2012. V. 33. S. 59.
81. Mejia M. a kol. // Hrudník. 2009. V. 136. S. 10.
82. Cottin V. a kol. //Eur. Respira. J. 2010. V. 35. S. 105.
83. Fell C.D. a kol. // Clin. Chest Med. 2012. V. 33. S. 51.
84. Gagermeier J. a kol. // Hrudník. 2005. V. 128. S. 601S.
85. Farkaš L. a kol. //Dopom. J. Respira. Cell Mol. Biol. 2011. V. 45. S. 1.
86. Kolilekas L. a kol. // J. Clin. Sleep Med. 2013. V. 9. S. 593.
87. Glaser S. a kol. // PLoS One. 2013. V. 8. P. e65643.
88. Žisman D.A. a kol. // N. Engl. J. Med. 2010. V. 363. S. 620.
89. Kráľ T.E. a kol. //Dopom. J. Respira. Crit. Care Med. 2011. V. 184. S. 92.
90. Raghu G. a kol. //Ann. Stážista. Med. 2013. V. 158. S. 641.
91. Raghu G. a kol. //Eur. Respira. J. 2013. V. 42. S. 1622.
92. Raghu G. a kol. //Eur. Respira. J. 2006. V. 27. S. 136.
93. Tobin R.W. a kol. //Dopom. J. Respira. Crit. Care Med. 1998. V. 158. S. 1804.
94. Sladké M.P. a kol. // J. Thorac. Cardiovasc. Surg. 2007. V. 133. S. 1078.
95. Savarino E. a kol. //Eur. Respira. J. 2013. V. 42. S. 1322.
96. Lee J.S. a kol. //Eur. Respira. J. 2012. V. 39. S. 352.
97. Lee J.S. a kol. //Dopom. J. Respira. Crit. Care Med. 2011. V. 184. S. 1390.
98. Lee J.S. a kol. // Lancet. Respira. Med. 2013. V. 1. S. 369.
99. Linden P.A. a kol. // J. Thorac. Cardiovasc. Surg. 2006. V. 31. S. 438.
100. Davis R.D. a kol. // J. Thorac. Cardiovasc. Surg. 2003. V. 125. S. 533.
101. Cantu E. III a kol. //Ann. Thorac. Surg. 2004. V. 78. S. 1142.
102. Hubbard R. a kol. //Dopom. J. Respira. Crit. Care Med. 2000. V. 161. S. 5.
103. Aubry M.C. a kol. // Mayo Clin. Proc. 2002. V. 77. S. 763.
104. Daniels C.E. a kol. //Curr. Opin. Pulm. Med. 2005. V. 11. S. 431.
105. Le Jeune I. a kol. // Respira. Med. 2007. V. 101. S. 2534.
106. Harris J.M. a kol. // Hrudník. 2010. V. 65. S. 70.
107. Kishi K. a kol. // J. Výpočet. Asistencia. Tomogr. 2006. V. 30. S. 95.
108. Yoshida R. a kol. // A.J.R. Am. J. Roentgenol. 2012. V. 199. S. 85.
109. Kushibe K. a kol. // Hrudník. Cardiovasc. Surg. 2007. V. 55. S. 505.
110. Park J.S. a kol. // Hrudník. Cardiovasc. Surg. 2011. V. 59. S. 148.
111. Chambers R.C. a kol. //Proc. Am. Thorac. Soc. 2012. V. 9. S. 96.
112. Hubbard R.B. a kol. //Dopom. J. Respira. Crit. Care Med. 2008. V. 178. S. 1257.
113. Sode B.F. a kol. //Dopom. J. Respira. Crit. Care Med. 2010. V. 181. S. 1085.
114. Sprunger D.B. a kol. //Eur. Respira. J. 2012. V. 39. S. 125.
ATMOSFÉRA
Na webovej stránke -preSS.ru si môžete ZAKÚPIŤ všetky naše knihy, časopisy a CD
za ceny vydavateľa bez obchodných prirážok.
Aj na webovej stránke atm-preSS.ru S BEZPLATNÝM PRÍSTUPOM nájdete archívy časopisov "Praktická pneumológia", "Atmosféra. Pulmonológia a alergológia", "Astma a alergia", "Aktuality o atmosfére. Kardiológia", "Nervové choroby", "Nervy", "Medicína", preklady manuálov a brožúr do ruštiny.
3740 0
Dr Toby Maher, vedecký pracovník Národného inštitútu pre výskum zdravia v Spojenom kráľovstve, odborný lekár v Royal Brompton Hospital v Londýne
Idiopatická pľúcna fibróza je progresívne ochorenie neznámeho pôvodu, ktoré je charakterizované postupným zjazvením, náhradou zdravého pľúcneho tkaniva s nevyhnutným výsledkom pľúcneho zlyhania.V našom dnešnom článku budeme hovoriť o idiopatickej pľúcnej fibróze, jej diagnostike a liečbe, ako aj o vyhliadkach na boj proti tejto chorobe.
Dr Toby Maher je výskumným pracovníkom Národného inštitútu pre výskum zdravia Spojeného kráľovstva a konzultantom v nemocnici Royal Brompton Hospital (Londýn). Prednáša na Imperial College London.
Doktor Maher je špecialistom na intersticiálnu chorobu pľúc a sarkoidózu.
Medzi jeho výskumné záujmy patrí vývoj nových biomarkerov pre pľúcne ochorenia, klinické skúšky nových liekov a štúdium patogenézy idiopatickej pľúcnej fibrózy (IPF).
Dr. Maher predtým pôsobil ako šéfredaktor Respirology a bol redaktorom PLOS One. Je členom redakčnej rady prestížneho časopisu Lancet Respiratory Medicine. Autor viac ako stoviek článkov a publikácií.
- Dr. Maher, čo je idiopatická pľúcna fibróza?
- Idiopatická pľúcna fibróza (IPF) je ťažké, smrteľné ochorenie, ktoré postihuje 3 milióny ľudí na celom svete.Hoci pľúcna fibróza zabije každý rok viac ľudí ako niektoré druhy rakoviny, toto ochorenie často prehliadajú aj lekári a vedci vedia o IPF prekvapivo málo.
Pri IPF dochádza k postupnému zjazveniu a znižuje sa funkcia výmeny plynov v pľúcach. Ako choroba postupuje, orgány a tkanivá dostávajú stále menej kyslíka a vzniká respiračné zlyhanie.
Ak sa spočiatku dýchavičnosť objavuje len pri námahe, tak sa časom život pacientov s IPF stáva každodenným bojom. Aj tie najjednoduchšie úkony, ako je sprchovanie či obliekanie, si od nich vyžadujú nadľudské úsilie.
Rýchlosť progresie IPF je rôzna. V priemere každý rok 1 z 20 pacientov zažije katastrofálne zhoršenie choroby. Epizódy exacerbácie vyžadujú hospitalizáciu a intenzívnu liečbu: v 50 % prípadov exacerbácie IPF sú pacienti usmrtení do 30 dní.
Celkovo je prognóza idiopatickej pľúcnej fibrózy zlá. Priemerná dĺžka života bez liečby je 2-3 roky od okamihu diagnózy. Päťročná miera prežitia nepresahuje 20%; toto číslo je porovnateľné s pľúcnym adenokarcinómom.
- Pomáha včasná diagnostika IPF zlepšiť prognózu?
- Včasná presná diagnóza idiopatickej pľúcnej fibrózy je skutočne veľmi dôležitá: pacienti dostanú adekvátnu liečbu včas a dlhšie si udržia vysokú kvalitu života.Bohužiaľ, podobnosť medzi príznakmi IPF a inými, bežnejšími pľúcnymi ochoreniami (astma, CHOCHP) veľmi sťažuje diagnostiku. V polovici prípadov IPF sú pacienti na začiatku nesprávne diagnostikovaní.
V dôsledku toho je priemerný čas medzi objavením sa prvých príznakov idiopatickej pľúcnej fibrózy a diagnózou IPF približne 1-2 roky.
Dva stratené roky!
Celý ten čas pacienti neúspešne bojujú s neexistujúcim ochorením, kým sa neobrátia na špecializované centrum, ktoré má skúsenosti s diagnostikou intersticiálnych pľúcnych ochorení.
Rýchly prístup k takýmto centrám a odborníkom je rozhodujúci pre presnú diagnózu a včasné začatie správnej liečby IPF.
Musíme pochopiť, že idiopatická pľúcna fibróza je nevyliečiteľná choroba, takže na riešenie emocionálnych problémov, ktoré vznikajú po vypočutí diagnózy, sú potrební psychológovia.
Najnovší globálny prieskum o idiopatickej pľúcnej fibróze (IPF), ktorý zverejnila spoločnosť Boehringer Ingelheim, zistil, že 49 % pacientov po diagnostikovaní pociťuje „úzkosť“ a 45 % „strach“. Ich pocity môžu ovplyvniť životné rozhodnutia, preto je odborná pomoc takýmto pacientom nevyhnutná.
- Aká je liečba idiopatickej pľúcnej fibrózy? Ako môže moderná medicína pomôcť pacientom, ak je IPF nevyliečiteľná?
- Hoci pľúcnu fibrózu nemožno vyliečiť, existujú rôzne možnosti na spomalenie IPF, zmiernenie príznakov a zlepšenie kvality života.Patria sem antifibrotiká, kyslík, antitusiká a bronchodilatanciá, rehabilitačné opatrenia a paliatívna starostlivosť na konci života.
Až donedávna neboli k dispozícii žiadne nové lieky na liečbu IPF. Situácia sa zmenila s príchodom antifibrotických liekov pirfenidón a nintedanib v USA a EÚ. Tieto lieky môžu spomaliť progresiu ochorenia.
Neliečivé možnosti pomáhajú zlepšiť pohodu a kvalitu života pacientov. Program pľúcnej rehabilitácie je postavený na fyzickom cvičení a zahŕňa celý tím špecializovaných špecialistov, fyzioterapeutov.
Okrem zlepšovania fyzickej zdatnosti a tolerancie cvičenia informujeme pacientov o tom, ako žiť s IPF, čo môžu a čo nie, a podporujeme ich v ťažkých časoch.
Niekoľko veľkých štúdií potvrdilo, že pľúcna rehabilitácia dosahuje ciele a umožňuje pacientom viesť plnohodnotnejší život.
Ako som povedal, u 1 z 20 pacientov s IPF sa každý rok vážne zhoršia symptómy, čo vedie k hospitalizácii na lôžkovom mieste. V súčasnosti neexistujú spoľahlivé terapeutické možnosti, ktoré by spoľahlivo zlepšili výsledky takýchto kríz (zvyčajne podávame kortikosteroidy a antibiotiká).
- Ako vidíte budúcnosť liečby idiopatickej pľúcnej fibrózy?
- Za posledných pár rokov urobila veda veľký pokrok v pochopení patogenézy, klinického obrazu a sľubných cieľov liečby IPF.Dúfam, že budúcnosť prinesie dobré správy miliónom pacientov a ich rodinám.
Najdôležitejšie je, že sa čoraz viac chápe dôležitosť včasnej diagnostiky a liečby pľúcnej fibrózy. Vznikajú nové špecializované centrá, študuje nová generácia lekárov, ktorí rozumejú zložitosti IPF. V mnohých krajinách sa vytvára ucelený systém starostlivosti o takýchto pacientov.
Pozitívne zmeny a dôležitosť vedeckého výskumu si uvedomujú aj samotní pacienti.
Ten istý globálny prieskum spoločnosti Boehringer Ingelheim ukazuje, že 20 % pacientov s idiopatickou pľúcnou fibrózou (IPF) naďalej žije v nádeji na budúci pokrok v boji proti ich chorobe. Financovanie výskumu sa totiž postupne zvyšuje a úspech tejto politiky je evidentný už dnes.
Dnes všade prebiehajú klinické skúšky nových liekov, ktoré podávajú ruku nádeje ťažko chorým pacientom. Máme celý rad prebiehajúcich a plánovaných štúdií: nové lieky, kombinácie existujúcich liekov, diagnostické a terapeutické biomarkery.
: magister farmácie a odborný lekársky prekladateľ
Únava a znížená hladina kyslíka v krvi. Niekedy je pľúcna fibróza spôsobená látkami z vonkajšieho prostredia, ktoré sa dajú identifikovať. Ale v mnohých prípadoch zostáva príčina ochorenia nejasná. Ak je príčina pľúcnej fibrózy neznáma, stav sa nazýva idiopatická pľúcna fibróza (IPF); ochorenie sa predtým nazývalo idiopatická fibrózna alveolitída (IFA), ale tento termín sa už nepoužíva.
Čísla a fakty
- Neuskutočnili sa žiadne rozsiahle štúdie o incidencii a incidencii IPF.
- Podľa rôznych zdrojov trpí IPF 2 až 29 ľudí na 100 tisíc obyvateľov.
- Nie je známe, či geografické, etnické, kultúrne alebo rasové faktory ovplyvňujú výskyt a výskyt IPF.
- U väčšiny pacientov s IPF sa vo veku 50 až 70 rokov objavia príznaky ako kašeľ a dýchavičnosť. IPF je menej častá u ľudí mladších ako 50 rokov.
- Dlho sa predpokladalo, že IPF sa vyskytuje častejšie u mužov ako u žien, no v posledných rokoch došlo k nárastu výskytu IPF u žien.
- V niektorých prípadoch sa IPF vyvinie u viac ako jednej osoby z tej istej rodiny. Keď k tomu dôjde, choroba sa nazýva familiárna pľúcna fibróza. Skutočnosť, že pľúcna fibróza je niekedy dedičná, viedla mnohých odborníkov k presvedčeniu, že prítomnosť určitých génov môže viesť k rozvoju ochorenia.
Kedy navštíviť lekára
- Na suchý kašeľ alebo ťažkosti s dýchaním, ktoré sa časom nezlepšujú.
- Ak sa váš stav náhle zhorší a príznaky sa zhoršia, mali by ste okamžite vyhľadať pomoc.
Diagnóza ochorenia
Lekár môže mať podozrenie na IPF na základe symptómov, ako je kašeľ a ťažkosti s dýchaním. Patologické zvuky v pľúcach, nazývané crepitus, môže lekár počuť v momente hlbokej inšpirácie. Pacient a ošetrujúci lekár si môžu všimnúť zhrubnutie prstov na samotných koncoch a charakteristickú zmenu ich tvaru, takzvané paličky. Prítomnosť týchto príznakov je dôvodom na odoslanie pacienta k špecialistovi na pľúcne ochorenia.
Pneumológ vykoná kompletné vyšetrenie a môže si objednať niekoľko vyšetrení, ako je röntgen hrudníka, test respiračných funkcií (spirometria) alebo meranie hladiny kyslíka v krvi. Okrem toho môže byť potrebné vyšetrenie hrudníka pomocou počítačovej tomografie (HRCT) s vysokým rozlíšením, echokardiogram (ultrazvuk srdca) a niekedy môže byť potrebná biopsia pľúc.
Pľúcna biopsia sa zvyčajne vykonáva pomocou video-asistovanej torakoskopickej chirurgie (VATS - video assisted thoracoscopic surgery) v celkovej anestézii. Chirurg pri tomto zákroku urobí do hrudnej steny dva alebo tri malé otvory, cez ktoré zasunie videokameru na pružnú základňu. Prístroj umožňuje nahliadnuť do hrudnej dutiny a odobrať kúsok pľúcneho tkaniva na vyšetrenie.
Liečba choroby
Po diagnostikovaní IPF by pacienti mali pravidelne navštevovať pulmonológa. Liečba IPF je predovšetkým symptomatická, zameraná na zmiernenie kašľa a dýchavičnosti. Dva nové špecifické lieky na liečbu IPF, ktoré spomaľujú progresiu fibrózy, boli schválené na použitie v Spojených štátoch. Tieto lieky sú dostupné aj v Rusku, aj keď, bohužiaľ, náklady na lieky sú veľmi vysoké.
Pred príchodom špecifických liekov sa na liečbu IPF používali glukokortikosteroidné hormóny (kortikosteroidy) a imunosupresíva, ktoré však neboli veľmi účinné a spôsobovali mnohé nežiaduce vedľajšie účinky. Pľúcna rehabilitácia, oxygenoterapia a liečba pľúcnej hypertenzie sa tiež používajú na zmiernenie symptómov IPF a súvisiacich stavov.
Do práce s pacientom s IPF by sa malo zapojiť mnoho odborníkov: pneumológovia, fyzioterapeuti, špecialisti na paliatívnu starostlivosť, fyzioterapeuti. Mnohé z nich sa u nás ešte len začínajú objavovať. Porozprávajte sa so svojím lekárom o možných liekoch a terapiách, ktoré môžu pomôcť vo vašom konkrétnom prípade.
Transplantácia pľúc pre IPF
Transplantácia pľúc je dnes jediným spôsobom, ako zvýšiť dĺžku života u pacientov s IPF. Transplantácia je veľký chirurgický zákrok, ktorý si vyžaduje celoživotnú liečbu liekmi, ktoré bránia imunitnému systému odmietnuť darcovské pľúca. Nie všetci pacienti s IPF sú vhodní na transplantáciu pľúc. Ošetrujúci pneumológ môže zhodnotiť stav, aby určil, či je v konkrétnom prípade možná transplantácia. Toto hodnotenie môže trvať mesiace, takže lekár môže zvážiť transplantáciu pľúc skôr, ako sa stav začne zhoršovať.
Popredné inštitúcie vykonávajúce transplantáciu pľúc v Rusku sú Federálne vedecké centrum pre transplantológiu pomenované po ňom. Akademik V.I. Shumakov a Výskumný ústav SP pomenovaný po. N.V. Sklifosovský.
Pľúcna rehabilitácia
Zapojenie do programu pľúcnej rehabilitácie a účasť v podporných skupinách je nevyhnutná, aby ste sa dozvedeli viac o chorobe a možnostiach liečby. Programy pľúcnej rehabilitácie môžu oživiť a zlepšiť celkový tonus, znížiť dýchavičnosť, poskytnúť lepšie pochopenie IPF a používania kyslíka a naučiť sa zručnostiam starostlivosti o seba.
Saturácia krvi kyslíkom by sa mala vždy udržiavať nad 89%, bez ohľadu na to, čo človek robí: sedí, chodí, cvičí alebo spí. Ale ako choroba postupuje, potreba dodatočného kyslíka sa môže zmeniť. Preto je dôležité pravidelne hodnotiť hladinu kyslíka, aby ste pochopili, koľko kyslíka je v tomto štádiu dostatočné v pokoji, počas fyzickej aktivity alebo počas spánku.
Pre fajčiarov je veľmi dôležité s týmto zlozvykom skoncovať. Tabakový dym zhoršuje dýchacie problémy.
Preventívne opatrenia
Ak máte chronické ochorenie pľúc, je veľmi dôležité vyhnúť sa situáciám, v ktorých sa môžete nakaziť ARVI a chrípkou. Proti chrípke je potrebné sa dať zaočkovať každý rok. U malého percenta pacientov s IPF dôjde k náhlej exacerbácii stavu a dýchavičnosť v dôsledku IPF sa rýchlo zhoršuje. Nikto nevie, prečo dochádza k náhlym exacerbáciám a u ktorých pacientov je ich výskyt pravdepodobnejší. Ak spozorujete náhle zhoršenie dýchavičnosti, kontaktujte svojho poskytovateľa zdravotnej starostlivosti alebo vyhľadajte pohotovostnú lekársku pomoc.
Účasť na klinických štúdiách IPF
Ak máte záujem zúčastniť sa výskumu, opýtajte sa svojho pneumológa. Keď sú k dispozícii nové spôsoby liečby, uskutočňujú sa klinické štúdie s cieľom pochopiť, ako každá liečba funguje. Tieto štúdie sa môžu vykonávať len u dobrovoľníkov s IPF. Možno by stálo za to skontrolovať, či v niektorom z výskumných centier v blízkosti vášho bydliska prebieha výskum IPF. Aj keď nemáte v úmysle byť účastníkom výskumu, pomoc od centra, ktoré sa špecializuje na IPF, môže byť užitočné.
V roku 2017 bolo v Jekaterinburgu otvorené prvé Regionálne centrum pre diagnostiku pacientov s IPF.
Ako sa pripraviť na návštevu
Vopred si urobte zoznam svojich príznakov a otázok, ktoré by ste chceli prediskutovať so svojím lekárom. Je tiež dôležité zapamätať si (a zapísať si), kedy ste si prvýkrát všimli príznaky a ako sa časom menili. Je dobré, ak na stretnutie prídu vaši príbuzní, aby vám pomohli položiť ďalšie otázky alebo si zapamätať dôležité informácie.
Idiopatická pľúcna fibróza (kryptogénna fibrotizujúca alveolitída) je najbežnejšou formou idiopatickej intersticiálnej pneumónie, ktorá zodpovedá progresívnej pľúcnej fibróze a prevláda u mužov fajčiarov. Symptómy idiopatickej pľúcnej fibrózy sa vyvíjajú mesiace až roky a zahŕňajú dýchavičnosť pri námahe, kašeľ a sipot.
Diagnóza sa stanovuje na základe anamnézy, fyzikálneho vyšetrenia, röntgenového vyšetrenia hrudníka a testov funkcie pľúc a v prípade potreby sa potvrdzuje pomocou HRCT, pľúcnej biopsie alebo oboch. Žiadna špecifická liečba idiopatickej pľúcnej fibrózy nepreukázala účinnosť, ale často sa predpisujú glukokortikoidy, cyklofosfamid, azatioprín alebo ich kombinácie. U väčšiny pacientov dochádza k zhoršeniu dokonca aj pri liečbe; medián prežívania je menej ako 3 roky od diagnózy.
Kód ICD-10
J84.1 Iné intersticiálne pľúcne ochorenia so zmienkou o fibróze
Príčiny idiopatickej pľúcnej fibrózy
Idiopatická pľúcna fibróza, histologicky definovaná ako zvyčajná intersticiálna pneumónia, predstavuje 50 % prípadov idiopatickej intersticiálnej pneumónie a vyskytuje sa u mužov aj žien vo veku 50 až 60 rokov v pomere 2:1. Pokračovanie alebo predchádzajúce fajčenie silne koreluje s ochorením. Existuje určitá genetická predispozícia: v 3% prípadov sa pozoruje zaťažená rodinná anamnéza.
Hoci sa idiopatická pľúcna fibróza nazýva pneumónia, zápal pravdepodobne zohráva relatívne malú úlohu. Predpokladá sa, že environmentálne, genetické alebo iné neznáme faktory spočiatku spôsobujú poškodenie alveolárneho epitelu, ale proliferácia špecifických a aberantných intersticiálnych fibroblastov a mezenchymálnych buniek (s ukladaním kolagénu a fibrózou) je pravdepodobne základom klinickej progresie ochorenia. Kľúčovými histologickými kritériami sú subpleurálna fibróza s oblasťami proliferácie fibroblastov a oblasťami závažnej fibrózy rozptýlenými oblasťami normálneho pľúcneho tkaniva. Rozsiahly intersticiálny zápal je sprevádzaný lymfocytárnou, plazmocytickou a histiocytárnou infiltráciou. Cystická dilatácia periférnych alveol („plástové pľúca“) sa nachádza u všetkých pacientov a zvyšuje sa s progresiou ochorenia. Táto histologická štruktúra je pri IBLAP známej etiológie neobvyklá; Termín zvyčajná intersticiálna pneumónia sa používa pre idiopatické lézie, ktoré nemajú zjavnú príčinu.
Príznaky idiopatickej pľúcnej fibrózy
Symptómy idiopatickej pľúcnej fibrózy sa zvyčajne rozvíjajú počas 6 mesiacov až niekoľkých rokov a zahŕňajú dýchavičnosť pri námahe a neproduktívny kašeľ. Celkové príznaky (horúčka až subfebrilné hladiny a myalgia) sú zriedkavé. Klasickým znakom idiopatickej pľúcnej fibrózy je hlasné, suché bilaterálne bazálne inspiračné praskanie (pripomínajúce zvuk otvárania suchého zipsu). Zhrubnutie koncových falangov prstov je prítomné približne v 50 % prípadov. Ostatné vyšetrovacie nálezy zostávajú v norme, kým sa nevyvinie konečné štádium ochorenia, kedy sa môžu rozvinúť prejavy pľúcnej hypertenzie a systolickej dysfunkcie pravej komory.
Diagnóza idiopatickej pľúcnej fibrózy
Diagnóza je založená na rozbore anamnestických údajov, výsledkov rádiologických vyšetrení, testov funkcie pľúc a biopsií. Idiopatická pľúcna fibróza je zvyčajne nesprávne diagnostikovaná ako iné ochorenia s podobnými klinickými prejavmi, ako je bronchitída, bronchiálna astma alebo srdcové zlyhanie.
Liečba idiopatickej pľúcnej fibrózy
Žiadna zo špecifických možností liečby nepreukázala účinnosť. Udržiavacia liečba idiopatickej pľúcnej fibrózy je obmedzená na inhaláciu kyslíka pri hypoxémii a antibiotiká na rozvoj pneumónie. Konečné štádium ochorenia môže u vybraných pacientov vyžadovať transplantáciu pľúc. Glukokortikoidy a cytotoxické látky (cyklofosfamid, azatioprín) sa tradične empiricky podávajú pacientom s idiopatickou pľúcnou fibrózou v snahe zastaviť progresiu zápalu, ale len obmedzené údaje naznačujú ich účinnosť. Bežnou praxou je však vyskúšať prednizolón (perorálne, v dávke 0,5 mg/kg až 1,0 mg/kg, jedenkrát denne počas 3 mesiacov, po čom nasleduje zníženie dávky na 0,25 mg/kg, jedenkrát denne počas nasledujúceho 3-6 mesiacov) v kombinácii s cyklofosfamidom alebo azatioprínom (perorálne, v dávke 1 mg/kg až 2 mg/kg, 1-krát denne a N-acetylcysteín 600 mg 3-krát denne perorálne ako antioxidant). V intervaloch každé 3 mesiace až 1-krát za rok sa vykonáva klinické, rádiologické a fyzikálne hodnotenie stavu a úprava dávky liekov. Liečba idiopatickej pľúcnej fibrózy sa preruší, ak nedôjde k objektívnej odpovedi.
Pirfenidón, látka, ktorá inhibuje syntézu kolagénu, môže stabilizovať funkciu pľúc a znížiť riziko exacerbácií. Účinnosť iných antifibrotických činidiel, najmä tých, ktoré inhibujú syntézu kolagénu (relaxín), profibrotické rastové faktory (suramín) a endotelín-1 (blokátor receptora angiotenzínu) bola preukázaná iba in vitro.
Interferón-y-lb preukázal dobrý účinok, keď sa podával s prednizolónom v malej štúdii, ale veľká dvojito zaslepená medzinárodná randomizovaná štúdia nepreukázala žiadny vplyv na prežívanie bez ochorenia, funkciu pľúc alebo kvalitu života.
M.Yu Brovko, L.A. Akulkina, V.I. Šolomová, M.V. Lebedeva
Idiopatická pľúcna fibróza (IPF) je variant idiopatickej intersticiálnej pneumónie (IIP) charakterizovaný neúprosne progresívnym priebehom a vysokou mortalitou. Na rozdiel od väčšiny IIP imunosupresívna liečba neovplyvňuje rýchlosť progresie IPF. V poslednom desaťročí bola preukázaná účinnosť dvoch antifibrotických liekov v liečbe IPF – pirfenidónu a nintedanibu. Pre včasné začatie patogenetickej liečby je potrebné čo najrýchlejšie stanoviť diagnózu IPF na základe diagnostického algoritmu, ktorý zahŕňa analýzu klinických, laboratórnych a inštrumentálnych údajov, predovšetkým výsledkov počítačovej tomografie s vysokým rozlíšením. (HRCT). Ak nie je dostatočne informatívna, možno použiť minimálne invazívnu transbronchiálnu kryobiopsiu pľúc, ktorá je presnosťou porovnateľná s chirurgickou pľúcnou biopsiou. Hľadanie molekulárnych biologických a genetických markerov IPF pokračuje.
ObsahKlasifikácia American Thoracic Society/European Respiratory Society (ATS):1–112. /ERS):1–112.), idiopatická pľúcna fibróza (IPF) je formou idiopatickej intersticiálnej pneumónie (IIP) (tabuľka 1). Podiel IPF v štruktúre všetkých IIP je 20 – 30 % a incidencia je od 7 do 17 prípadov na 100 000 obyvateľov. Muži ochorejú o niečo častejšie ako ženy (pomer muži/ženy približne 1,5:1). IPF sa vyvíja najmä u ľudí v strednom a staršom veku: 65 % pacientov má v čase diagnózy 60 rokov a viac.
| Bežné formy IIP |
| Idiopatická pľúcna fibróza (IPF) |
| Idiopatická nešpecifická intersticiálna pneumónia |
| Respiračná bronchiolitída spojená s intersticiálnym ochorením pľúc |
| Deskvamatívna intersticiálna pneumónia |
| Kryptogénna organizujúca sa pneumónia |
| Akútna intersticiálna pneumónia |
| Zriedkavé formy IIP |
| Idiopatická lymfocytová intersticiálna pneumónia |
| Idiopatická pleuroparenchymálna fibroelastóza |
| Neklasifikovateľné formy (IIP) |
V roku 2018 P. Wolters a kol. navrhol rozlíšiť 4 varianty pľúcnej fibrózy v závislosti od patogenézy ochorenia (tabuľka 2). IPF sa vyznačuje progresívnym priebehom s rozvojom respiračného zlyhania a má najnepriaznivejšiu prognózu spomedzi všetkých IIP: priemerná miera prežitia je od 2 do 5 rokov. Vysoká úmrtnosť pacientov s IPF sa vysvetľuje zvláštnosťami patogenézy ochorenia – prevahou fibrózy s miernou závažnosťou zápalových zmien. Hlavným mechanizmom vedúcim k rozvoju progresívnej pľúcnej fibrózy je pretrvávajúce poškodenie alveolárneho epitelu s následným narušením jeho regeneračných procesov, nadmerné ukladanie zložiek extracelulárnej matrice a aktivácia fibroblastov a myofibroblastov. Tieto zmeny určujú neúčinnosť tradičnej imunosupresívnej liečby u pacientov s IPF. V súčasnosti však došlo k výraznému pokroku v liečbe IPF spojenej s užívaním antifibrotických liekov – pirfenidónu (transformujúci rastový faktor beta – antagonista TGF beta) a nintedanibu (viacnásobný inhibítor tyrozínkinázy), ktoré spomaľujú pokles pľúcnych objemov, primárne vynútenú dĺžku života, kapacitu pľúc (FVC) a zlepšiť prežívanie bez progresie. Pri absencii kontraindikácií sa transplantácia pľúc zvažuje aj ako možnosť liečby u pacientov s pokročilou IPF komplikovanou ťažkým respiračným zlyhaním.
| Skupina 1: LF indukovaná dysfunkciou epitelových buniek | ILF |
| Skupina 2: LF indukovaná dysfunkciou zápalových buniek | Systémová sklerodermia, reumatoidná artritída, Sjogrenov syndróm, exogénna alergická alveolitída, sarkoidóza, NSIP |
| Skupina 3: LF spôsobená užívaním liekov alebo vystavením pracovným faktorom | Azbestóza, silikóza, poškodenie pľúc vyvolané liekmi |
| Skupina 4: DF súvisiaca s fajčením | Deskvamatívna intersticiálna pneumónia, respiračná bronchiolitída spojená s intersticiálnou chorobou pľúc, histiocytóza z Langerhansových buniek |
Klinický obraz
Hlavnými ťažkosťami u pacientov s IPF sú progresívna dýchavičnosť a suchý kašeľ, ktoré sa zhoršujú fyzickou aktivitou. Menej časté sú bolesti a nepríjemné pocity na hrudníku, zvýšená únava, celková slabosť a strata hmotnosti. V niektorých prípadoch je ochorenie v počiatočných štádiách asymptomatické a prvými prejavmi sú zmeny funkčných pľúcnych parametrov. Typickým auskultačným fenoménom pri IPF je krepitus, prevažne v posterobazálnych oblastiach pľúc. U pacientov s pokročilým štádiom IPF sa môžu vyskytnúť známky sekundárnej arteriálnej pľúcnej hypertenzie s rozvojom cor pulmonale a srdcového zlyhania pravej komory.
Pri IPF sa môže zistiť mierne zvýšenie ESR. Napriek prítomnosti progresívneho respiračného zlyhania je výrazné zvýšenie koncentrácie hemoglobínu extrémne zriedkavé. zníženie všetkých objemov pľúc v kombinácii so znížením difúznej kapacity pľúc (DLCO). Jedným zo skorých prejavov IPF môže byť izolovaný pokles DLCO s relatívnym zachovaním pľúcnych objemov. Včasné prejavy IPF tiež zahŕňajú zvýšenie alveoloarteriálneho kyslíkového gradientu, ktorý je často charakterizovaný normálnou saturáciou krvi v pokoji a desaturáciou počas cvičenia.
Diagnostický algoritmus
Diagnóza IPF je založená na absencii známej príčiny pľúcnej fibrózy a na prítomnosti vzoru obvyklej intersticiálnej pneumónie (UIP). Dokonca aj v prítomnosti histologického obrazu AIP pri chirurgickej pľúcnej biopsii (SBL) si konečná diagnóza vyžaduje vylúčenie iných patologických stavov spojených s rozvojom AIP, vrátane difúznych ochorení spojivového tkaniva, pneumokoniózy, ochorenia pľúc súvisiaceho s liekmi a familiárna pľúcna fibróza. Pri absencii údajov pre alternatívnu diagnózu sa podľa súčasných klinických odporúčaní stanovuje diagnóza IPF na základe charakteristických nálezov počítačovej tomografie s vysokým rozlíšením (HRCT) a v prípade potreby na základe výsledkov biopsie pľúc (tab. 3). Treba poznamenať, že v predloženej histologickej klasifikácii sa rozlišuje „možná IPF“ a „pravdepodobná IPF“, keď nie je možné jednoznačne potvrdiť alebo vylúčiť prítomnosť IPF. V tomto prípade je na objasnenie diagnózy indikované prehodnotenie údajov HRCT a pľúcnej biopsie.
| CT snímka | Histologické údaje | Diagnóza |
|---|---|---|
| OIP | OIP | ILF |
| Pravdepodobný AIP | ||
| Možné OIP | ||
| Neklasifikovaná fibróza | ||
| Nezodpovedá OIP | Non-IPF | |
| Možné OIP | OIP Pravdepodobný OIP | ILF |
| Možné OIP | Pravdepodobný IPF | |
| Neklasifikovaná fibróza | ||
| Nezodpovedá OIP | Non-IPF | |
| Nezodpovedá OIP | OIP | Možné IPF |
| Pravdepodobný AIP | Non-IPF | |
| Možné OIP | ||
| Neklasifikovaná fibróza | ||
| Nezodpovedá OIP |
CT diagnostika
HRCT hrá kľúčovú úlohu v diagnostike IPF a umožňuje diagnostiku približne v 2/3 prípadov. Viaceré štúdie ukázali, že CT obraz typickej AIP podľa HRCT je v súlade s prítomnosťou histologického obrazu typickej AIP podľa pľúcnej biopsie v 90 – 100 % prípadov. Prítomnosť spoľahlivých CT znakov UIP sa v súčasnosti považuje za dostatočnú na diagnostiku IPF bez pľúcnej biopsie. Chirurgická pľúcna biopsia (SLB) sa odporúča v prítomnosti CT obrazu, ktorý nie je typický pre AIP. V takýchto prípadoch sa diagnóza stanovuje na základe kombinácie údajov HRCT a histologických nálezov (tabuľka 3). Presná interpretácia údajov HRCT je teda predpokladom diagnózy.
V súčasnosti existujú tri varianty CT UIP: „typický UIP“, ktorý vylučuje potrebu CBL, „možný UIP“ a „nezodpovedá UIP“. Ak sú k dispozícii posledné dve možnosti, vyžaduje sa CBL.
CT vzhľad typickej AIP zahŕňa prevažne bazálne a periférne retikulárne zmeny s plástovosťou s trakčnou bronchiektáziou alebo bez nej. Za kritériá pre „voštinové pľúca“ sa považujú prevažne subpleurálne cysty s priemerom 3 – 10 mm s priehľadnými, relatívne hrubými stenami (1 – 3 mm), usporiadanými vo vrstvách. Všetky znaky CT považované za „nekonzistentné“ s UIP by nemali chýbať (obr. 1). Ak sú splnené všetky vyššie uvedené kritériá, údaje z HRCT sú dostatočné na diagnostiku AIP a nie je potrebná pľúcna biopsia. Pokiaľ ide o znaky typického AIP, závery rôznych špecialistov sú zvyčajne v dobrej zhode. Je však potrebné poznamenať, že AIP a IPF nie sú synonymá, pretože zmeny CT charakteristické pre AIP možno pozorovať pri množstve iných ochorení, predovšetkým pri difúznych ochoreniach spojivového tkaniva.
Ryža. 1. CT obraz typického AIP u 77-ročnej ženy.Pri možnej AIP sa pozorujú prevažne bazálne a periférne retikulárne zmeny bez tvorby plástových pľúcnych zón. V tomto prípade nedochádza k zmenám, ktoré by nezodpovedali OIP (obr. 2). Obraz možného AIP je pre IPF menej špecifický ako obraz typického AIP. V tomto prípade by sa mala diferenciálna diagnostika vykonať predovšetkým s nešpecifickou intersticiálnou pneumóniou (NSIP), ktorá je charakterizovaná absenciou oblastí voštinových pľúc, prevahou zákalových opacít nad retikulárnymi zmenami a relatívnou zachovanosťou subpleurálnych zón. Oblasti bunkovej transformácie sú v NSIP zriedkavé. V jednej štúdii sa našli u menej ako 5 % pacientov s idiopatickou NSIP.
 Ryža. 2. CT obraz možnej UIP u 75-ročného muža.(A) Na axiálnom reze sa zisťujú retikulárne zmeny, trakčné bronchiektázie a zóny „plástových pľúc“. (B) Koronálna rekonštrukcia ukazuje apikobazálny gradient poškodenia pľúcneho tkaniva.
Ryža. 2. CT obraz možnej UIP u 75-ročného muža.(A) Na axiálnom reze sa zisťujú retikulárne zmeny, trakčné bronchiektázie a zóny „plástových pľúc“. (B) Koronálna rekonštrukcia ukazuje apikobazálny gradient poškodenia pľúcneho tkaniva. Zmeny podľa údajov HRCT, ktoré sa nepovažujú za konzistentné s AIP, zahŕňajú: a) prevahu zmien v hornej a strednej časti pľúc; b) prevažne peribronchovaskulárne zmeny; c) významné oblasti stmavnutia typu „matné sklo“, ktorých prevalencia prevyšuje retikulárne zmeny; d) obojstranné ložiskové zmeny, hlavne v horných častiach pľúc; e) prítomnosť cýst (viacnásobných, bilaterálnych) mimo zón fibrózy; f) vzor mozaikového stmavnutia pľúcneho tkaniva/prítomnosť „lapačov vzduchu“ (bilaterálne zmeny v troch alebo viacerých lalokoch); g) prítomnosť konsolidačných zón (obr. 3).
 Obr.3. CT obraz, ktorý nezodpovedá AIP u 61-ročného pacienta s chronickou exogénnou alergickou alveolitídou. Na axiálnom reze sú viditeľné retikulárne zmeny v kombinácii s mozaikovým rozložením tmavších zón typu „brúseného skla“.
Obr.3. CT obraz, ktorý nezodpovedá AIP u 61-ročného pacienta s chronickou exogénnou alergickou alveolitídou. Na axiálnom reze sú viditeľné retikulárne zmeny v kombinácii s mozaikovým rozložením tmavších zón typu „brúseného skla“. Napriek vysokej pravdepodobnosti IPF pri typickej AIP podľa HRCT by absencia charakteristického CT obrazu nemala slúžiť ako základ na vylúčenie diagnózy IPF. V roku 2017 D. Lynch a spol. navrhol novú CT klasifikáciu AIP, v ktorej bola po prvýkrát identifikovaná skupina neurčitých AIP (tab. 4).
| Typické OIP | Pravdepodobný AIP | Neisté OIP | Najmenej pravdepodobne zodpovedá OIP |
|---|---|---|---|
| Prevaha v bazálnej a subpleurálnej oblasti (zriedkavo difúzne zmeny); často heterogénna distribúcia bunkovej zóny pľúc; retikulárne zmeny s periférnou trakčnou bronchoektáziou a bronchiolektázou; nedostatok údajov pre alternatívnu diagnózu | Prevaha v bazálnej a subpleurálnej oblasti; často heterogénna distribúcia Retikulárne zmeny s periférnou trakčnou bronchiektáziou a bronchiolektázou; absencia zón „bunkových pľúc“; nedostatok údajov pre alternatívnu diagnózu | Variabilná alebo difúzna distribúcia Prítomnosť fibrózy v kombinácii s malými zmenami objemu, ktoré nezodpovedajú AIP | Prevaha v hornej a strednej časti pľúc; peribronchovaskulárna distribúcia s relatívnym šetrením subpleurálnych zón Ktorýkoľvek z nasledujúcich: prevaha konsolidačných zón; Významne veľké oblasti zákalu (pri absencii exacerbácie IPF); difúzne fokálne alebo cystické zmeny; výrazné mozaikové stmavnutie pľúcneho tkaniva s prítomnosťou „lapačov vzduchu“ |
Klinický priebeh IPF sa môže líšiť. Väčšina pacientov má pomaly progresívny priebeh, ale niektorí pacienti majú stabilizáciu patologického procesu, zatiaľ čo iní majú pomerne rýchlu progresiu ochorenia. Čo sa týka závažnosti pľúcnych zmien podľa HRCT, zákalové zákalové zóny sa najčastejšie premieňajú na retikulárne zmeny, ktoré následne môžu progredovať a vytvárať plástové pľúcne zóny, ktorých veľkosť sa časom zvyčajne zväčšuje. Treba poznamenať, že všeobecný vzorec pľúcnych zmien sa môže tiež zmeniť: napríklad CT obraz možnej AIP sa môže transformovať na typickú AIP.
Biopsia pľúc
Ak nie je jednoznačný dôkaz prítomnosti IPF na HRCT, potom na potvrdenie diagnózy je indikovaná chirurgická pľúcna biopsia, ktorá sa často vykonáva pomocou videoasistovanej torakoskopickej techniky. Na zvýšenie účinnosti by sa mali pľúcne biopsie vykonávať z rôznych lalokov pľúc. Hoci je CBL najspoľahlivejšou metódou na stanovenie histologického obrazu IIP, je spojená s rizikom množstva komplikácií, z ktorých najzávažnejšou je exacerbácia IPF, najmä u pacientov s ťažkým respiračným a/alebo srdcovým zlyhaním. V tomto ohľade by sa rozhodnutie o jeho vykonaní malo prijať individuálne, berúc do úvahy klinický obraz, možné prínosy pre stanovenie presnej diagnózy, ako aj súhlas pacienta.
V poslednom desaťročí bola vyvinutá technika transbronchiálnej kryobiopsie pľúc (TBLC) na histologické potvrdenie diagnózy IPF a iných variantov IIP. Jeho hlavnými výhodami sú minimálne invazívnosť, bez potreby intubácie a inhalačnej anestézie a v dôsledku toho nízky výskyt komplikácií v kombinácii s možnosťou získania veľkého objemu pľúcnej biopsie, postačujúcej v drvivej väčšine prípadov na histologickú verifikáciu diagnózy. U pacientov bez typického obrazu AIP podľa HRCT údajov teda TBCL umožnila stanoviť diagnózu približne v 2/3 prípadov, čo je porovnateľné s účinnosťou CBL v podobnej situácii. TBCBL sa zároveň vyznačuje nižším rizikom perioperačných komplikácií (najčastejšie sa zaznamenáva rozvoj pneumotoraxu a život ohrozujúceho krvácania v mieste biopsie) a úmrtím, kratšou dobou hospitalizácie, čo umožňuje vykonávať TBCBL u pacientov s vysokou úrovňou anestetického rizika a prítomnosťou kontraindikácií na CBL Zavedenie TBCBL do klinickej praxe teda môže rozšíriť indikácie pre pľúcnu biopsiu a zvýšiť diagnostickú presnosť algoritmu na vyšetrenie pacientov s podozrením na IPF.
V morfologickej štúdii pacientov s podozrením na IPF G. Raghu et al. Existuje päť možných histologických profilov ochorenia (tabuľka 5). V kombinácii s rádiologickým nálezom sa používajú na potvrdenie/vylúčenie diagnózy IPF (tab. 3).
Odlišná diagnóza
Pacienti s podozrením na IPF by mali mať starostlivú diferenciálnu diagnostiku. Pri zistení CT obrazu, ktorý zodpovedá pravdepodobnému alebo možnému AIP, čo sa stáva pomerne často, by diferenciálna diagnóza mala zahŕňať predovšetkým chronickú exogénnu alergickú alveolitídu a fibrotický variant NSIP. U niektorých pacientov sa však CBL odporúčaná v tomto prípade nevykonáva z dôvodu prítomnosti kontraindikácií (závažné respiračné zlyhanie, sprievodné ochorenia, vekové obmedzenia) alebo neochoty pacienta.
Pri diferenciálnej diagnostike je tiež dôležité vylúčiť pľúcne poškodenie ako súčasť systémového ochorenia spojiva, najmä reumatoidnú artritídu, systémovú sklerodermiu, dermatomyozitídu, Sjogrenov syndróm, a to aj v prítomnosti CT obrazu typického AIP. Ak má pacient individuálne klinické prejavy alebo zvýšené hladiny laboratórnych autoimunitných markerov, ktoré nezodpovedajú konkrétnemu systémovému ochoreniu spojivového tkaniva, je možné stanoviť diagnózu intersticiálnej pneumónie s autoimunitnými znakmi.
Genetické markery IPF
V súčasnosti bolo identifikovaných množstvo mutácií a polymorfizmov génov podieľajúcich sa na remodelácii pľúcneho tkaniva a regulácii vrodenej a získanej imunity, spojených so vznikom IPF. Patria sem najmä mutácie v génoch kódujúcich povrchovo aktívne proteíny A a D (S): 1–112. P-A a S):1–112. PD), opísané u familiárnych foriem IPF. Množstvo štúdií odhalilo súvislosť genetických polymorfizmov s prognózou ochorenia: najmä prítomnosť jednotlivých jednonukleotidových polymorfizmov v géne TLR-3 (Toll-like receptor typ 3) je spojená s rýchlejšou progresiou choroba. V IPF bolo tiež opísaných množstvo polymorfizmov v génoch mucínu 5B (MUC5B) a TOLLIP (proteín interagujúci s Toll-like receptorom). Hoci štúdium genetických polymorfizmov nie je súčasťou diagnostického algoritmu IPF, pokračuje sa v hľadaní genetických markerov, ktoré môžu slúžiť ako prediktory rôznych variantov priebehu ochorenia a odpovede na liečbu.
Exacerbácia IPF
Exacerbácia IPF je závažný život ohrozujúci stav prejavujúci sa rýchlym nárastom respiračného zlyhania u pacientov s predtým stanovenou diagnózou IPF. Spravidla sa vyznačuje mimoriadne ťažkým priebehom; Úmrtnosť v niektorých štúdiách dosiahla 85 %. Na rozdiel od stabilného alebo pomaly progresívneho priebehu IPF sú diagnostické kritériá pre jej exacerbáciu menej jasne definované. Podľa N. Collarda a kol. Kritériá pre exacerbáciu IPF zahŕňajú prítomnosť predchádzajúcej alebo novodiagnostikovanej IPF s prudkým nárastom dýchavičnosti, rozvoj respiračného zlyhania počas predchádzajúcich 30 dní bez preukázanej príčiny, ako aj objavenie sa nových oblastí opacifikácie pľúcne tkanivo typu „brúsené sklo“ a/alebo konsolidácia na pozadí existujúcich skorších zmien zodpovedajúcich AIP – zóny retikulárnych zmien a „plástové pľúca“ (obr. 4). Vyššie uvedené kritériá však majú nízku špecificitu, a preto pri podozrení na exacerbáciu IPF treba vykonať diferenciálnu diagnostiku infekčného procesu, tromboembólie pľúcnej artérie a jej vetiev, pneumotoraxu, ako aj akútneho zlyhania ľavej komory s rozvoj pľúcneho edému.
 Ryža. 4. Multifokálny variant exacerbácie IPF. Podľa údajov HRCT je stmavnutie pľúcneho tkaniva určené typom „brúseného skla“ a konsolidáciou v centrálnej a periférnej časti pľúc v kombinácii so subpleurálnymi zmenami typu „plástové pľúca“.
Ryža. 4. Multifokálny variant exacerbácie IPF. Podľa údajov HRCT je stmavnutie pľúcneho tkaniva určené typom „brúseného skla“ a konsolidáciou v centrálnej a periférnej časti pľúc v kombinácii so subpleurálnymi zmenami typu „plástové pľúca“. Záver
Nástup nových liečebných postupov, najmä antifibrotických liekov, a zlyhanie tradičnej imunosupresívnej liečby IPF zdôrazňujú dôležitosť včasnej diagnózy a zahájenia liečby. Počas posledného desaťročia sa dosiahol významný pokrok vo vývoji diagnostických algoritmov pre pacientov s IPF. Tomu napomohlo zlepšenie kvality zobrazovacích techník, lepšie pochopenie úlohy pľúcnej biopsie a vývoj histologických kritérií pre IPF. Všetky vyššie uvedené parametre by mal vyšetrovať multidisciplinárny tím špecialistov, čo je v súčasnosti štandardom diagnostiky IPF. Napriek dosiahnutému pokroku zostávajú v diagnostike IPF nevyriešené otázky, ktoré súvisia najmä s používaním invazívnych diagnostických metód, najmä chirurgickej pľúcnej biopsie. Je potrebné pokračovať v hľadaní molekulárno-biologických a genetických markerov IPF a vo vývoji minimálne invazívnych bioptických metód pre čo najskoršiu diagnostiku, stanovenie prognózy a vývoj liečebných stratégií IPF.
Použité zdroje
- Travis WD, Costabel U, Hansell DM a kol. Oficiálny americký Thoracic S): 1–112. spoločnosť/Európske dýchacie S):1–112. vyhlásenie spoločnosti: Aktualizácia medzinárodnej multidisciplinárnej klasifikácie idiopatických intersticiálnych pneumónií. Am J Respir Crit Care Med 2013;188:733–48.
- Fernández Pérez ER, Daniels CE, S):1–112. Chroeder DR a kol. Incidencia, prevalencia a klinický priebeh idiopatickej pľúcnej fibrózy populačná štúdia. Hrudník 2010;137:129–37.
- Raghu G, Weycker D, Edelsberg J, a kol. Výskyt a prevalencia idiopatickej pľúcnej fibrózy. Am J Respir Crit Care Med 2006;174:810–6.
- Raghu G, Collard HR, Egan JJ a kol. Oficiálna ATS): 1–112. /ERS):1–112. /JRS):1–112. /ALAT S):1–112. Idiopatická pľúcna fibróza: Pokyny pre diagnostiku a manažment založené na dôkazoch. Am J Respir Crit Care Med 2011;183:788–824.
- Wolters PJ, Blackwell TS):1–112. Eickelberg O, a kol. Čas na zmenu: je idiopatická pľúcna fibróza stále idiopatická a iba fibrotická? Lancet Respir Med 2018; 6:154–60.
- Kráľ TE, Tooze J, S):1–112. chwarz MI a spol. Predpovedanie prežitia pri idiopatickej pľúcnej fibróze: skórovací systém a model prežitia. Am J Respir Crit Care Med 2001; 164:1171-81.
- Fomin V.V., Popova E.N., Lebedeva M.V. Idiopatická pľúcna fibróza: blížime sa k všeobecne uznávaným štandardom diagnostiky a liečby? Farmateka 2012;5:10–4.
- Armanios M, Alder JK, Chen JJ-L a kol. S): 1–112. Hortove teloméry sú rizikovým faktorom pre idiopatickú pľúcnu fibrózu. Proc Natl Acad S):1–112. ci USA):1–112. A 2008;105:13051–6.
- Leslie KO. Idiopatická pľúcna fibróza môže byť ochorenie rekurentného, trakčného poškodenia periférie starnúcich pľúc: Zjednocujúca hypotéza týkajúca sa etiológie a patogenézy. Arch Pathol Lab Med 2012;136:591–600.
- Blackwell TS): 1–112. Tager AM, Borok Z, a kol. Budúce smery výskumu idiopatickej pľúcnej fibrózy - správa z workshopu NHLBI. Am J Respir Crit Care Med 2014;189:214–22.
- Sieť klinického výskumu idiopatickej pľúcnej fibrózy, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednizón, azatioprín a N-acetylcysteín na pľúcnu fibrózu. N Engl J Med 2012;366:1968–77.
- Raghu G, Rochwerg B, Zhang Y a kol. Oficiálna ATS): 1–112. /ERS):1–112. /JRS):1–112. /ALAT Pokyny pre klinickú prax: Liečba idiopatickej pľúcnej fibrózy. Aktualizácia usmernenia pre klinickú prax z roku 2011. Am J Respir Crit Care Med 2015; 192:e3–19.
- Yusen RD, S):1–112. hearton TH, Qian Y, a kol. Transplantácia pľúc v Spojených štátoch: 1–112. Tates, 1999-2008: S):1–112. špeciálna vlastnosť. Am J Transplant 2010;10:1047–68.
- Avdeev S.N. Idiopatická pľúcna fibróza: moderný koncept a prístupy k diagnostike. Praktická pneumológia 2014;4:16–23.
- American Thoracic S):1–112. spoločnosti. Idiopatická pľúcna fibróza: diagnostika a liečba. Vyhlásenie medzinárodného konsenzu. American Thoracic S):1–112. spoločnosť (ATS):1–112.) a Európska respiračná S):1–112. spoločnosť (ERS):1–112.). Am J Respir Crit Care Med 2000; 161:646-64.
- Martinez FJ, Flaherty K. Testovanie funkcie pľúc pri idiopatickej intersticiálnej pneumónii. Proc Am Thorac S):1–112. ok 2006;3:315–21.
- Cepika A-M, Marinic I, Morovic-Vergles J, et al. Účinok steroidov na frekvenciu regulačných T buniek a expresiu FOXP3 u pacienta so systémovým lupus erythematosus: dvojročné sledovanie. Lupus 2007;16:374–7.
- Baldi BG, Pereira CA, Rubin AS): 1–112. a kol. Najdôležitejšie pokyny Brazílskej hrudnej asociácie pre intersticiálne pľúcne ochorenia. J Bras Pneumol 2012;38(3):282-91.
- Vzel A.A., Vzel I.Yu. Idiopatická pľúcna fibróza: stav problému. Časopis modernej klinickej medicíny 2017;10:14–21.
- Hodnett PA, Naidich DP. Fibrózna intersticiálna choroba pľúc: Praktický prístup k diagnostike a liečbe založený na počítačovej tomografii s vysokým rozlíšením a prehľad literatúry. Am J Respir Crit Care Med 2013;188:141–9.
- Flaherty KR, Thwaite EL, Kazerooni EA a kol. Rádiologická verzus histologická diagnóza pri UIP a NS):1–112. IP: S):1–112. urvival implikácie. Thorax 2003;58:143–8.
- Paulo P, Torres S): 1–112. Rabahi MF a kol. Obvyklá intersticiálna pneumónia: typické, možné a „nekonzistentné“ vzory. J Bras Pneumol 2017;43:393–8.
- Travis WD, Hunninghake G, King TE a kol. Idiopatická nešpecifická intersticiálna pneumónia: Správa American Thoracic S):1–112. spoločenský projekt. Am J Respir Crit Care Med 2008;177:1338–47.
- Lynch DA, S): 1–112. Verzellati N, Travis WD a kol. Diagnostické kritériá pre idiopatickú pľúcnu fibrózu: A Fleischner S):1–112. Biela kniha spoločnosti. Lancet Respir Med 2017;6:138–53.
- Kolb M, S):1–112. hargall Y. Operácia pľúc pri intersticiálnej chorobe pľúc – bezpečný a užitočný postup? J Thorac Dis 2013;5:375–7.
- Bango-Alvarez A, Ariza-Prota M, Torres-Rivas H a kol. Transbronchiálna kryobiopsia pri intersticiálnej chorobe pľúc: skúsenosti v 106 prípadoch – ako na to. ERJ Open Res 2017;3:00148–2016.
- Lentz RJ, Christine Argento A, Colby TV a kol. Transbronchiálna kryobiopsia pre difúzne parenchymálne ochorenie pľúc: Najmodernejší prehľad procesných technológií, súčasných dôkazov a budúcich výziev. J Thorac Dis 2017;9:2186–203.
- Tomassetti S):1–112. , Wells AU, Costabel U, a kol. Bronchoskopická kryobiopsia pľúc zvyšuje diagnostickú dôveru v multidisciplinárnu diagnostiku idiopatickej pľúcnej fibrózy. Am J Respir Crit Care Med 2016;193:745–52.
- Baddini-Martinez J, Baldi BG, Costa CH a kol. Aktualizácia diagnostiky a liečby idiopatickej pľúcnej fibrózy. J Bras Pneumol 2015;41:454–66.
- Tsvetková O.A., Voronkova O.O. Moderný prístup k liečbe pacientov s idiopatickou pľúcnou fibrózou. Clinical Med 2017;95:281–5.
- Avdeev S.N. Idiopatická pľúcna fibróza: moderné prístupy k terapii. Praktická pneumológia 2015;1 22–31.
- Fischer A, Antoniou KM, Brown KK a kol. Oficiálny európsky respiračný S):1–112. spoločnosť/American Thoracic S):1–112. výskum spoločnosti: Intersticiálna pneumónia s autoimunitnými znakmi. Eur Respir J 2015;46:976–87.
- Brownell R, Kaminski N, Woodruff PG a kol. Presná medicína: Nová hranica v idiopatickej pľúcnej fibróze. Am J Respir Crit Care Med 2016;193:1213–8.
- Thomas AQ, Lane K, Phillips J a kol. Heterozygotnosť pre mutáciu génu povrchovo aktívneho proteínu C spojená s obvyklou intersticiálnou pneumonitídou a bunkovou nešpecifickou intersticiálnou pneumonitídou v jednom druhu. Am J Respir Crit Care Med 2002; 165:1322–8.
- O'Dwyer DN, Armstrong ME, Trujillo G a kol. Polymorfizmus Toll-like receptora 3 L412F a progresia ochorenia pri idiopatickej pľúcnej fibróze. Am J Respir Crit Care Med 2013;188:1442–50.
- Oldham JM, Ma S):1–112. F, Martinez FJ a kol. TOLLIP, MUC5B a odpoveď na N-acetylcysteín u jedincov s idiopatickou pľúcnou fibrózou. Am J Respir Crit Care Med 2015;192:1475–82.
- Collard HR, Ryerson CJ, Corte TJ a kol. Akútna exacerbácia idiopatickej pľúcnej fibrózy správa medzinárodnej pracovnej skupiny. Am J Respir Crit Care Med 2016;194:265–75.
- Hambly N, Cox G, Kolb M. Akútne exacerbácie idiopatickej pľúcnej fibrózy: ťažko definovateľné; ťažšie zvládnuteľné. Eur Respir J 2017;49:1700811.
- Collard HR, Moore BB, Flaherty KR a kol. Akútne exacerbácie idiopatickej pľúcnej fibrózy. Am J Respir Crit Care Med 2007;176:636–43. "
- Akira M, Kozuka T, Yamamoto S): 1–112. , S): 1-112. Akatani M. Nálezy počítačovej tomografie pri akútnej exacerbácii idiopatickej pľúcnej fibrózy. Am J Respir Crit Care Med 2008;178:372–8.
- Juarez MM, Chan AL, Norris AG, a kol. Akútna exacerbácia idiopatickej pľúcnej fibrózy - prehľad súčasných a nových farmakoterapií. J Thorac Dis 2015;7:499–519
 Slabé skloňovanie podstatných mien
Slabé skloňovanie podstatných mien Informácie o prezidentoch našej krajiny
Informácie o prezidentoch našej krajiny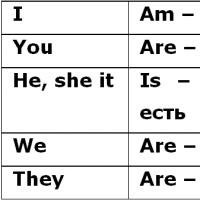 "Is", "are": použitie v angličtine Pravidlo tu bi v angličtine
"Is", "are": použitie v angličtine Pravidlo tu bi v angličtine Georgy Leonidovič Pyatakov: biografia Revolúcia a občianska vojna
Georgy Leonidovič Pyatakov: biografia Revolúcia a občianska vojna Musí prenajímateľ platiť daň z príjmu fyzických osôb z príjmu z prenájmu nehnuteľnosti?
Musí prenajímateľ platiť daň z príjmu fyzických osôb z príjmu z prenájmu nehnuteľnosti? Ako napísať vysvetlenie k žiadosti daňového inšpektorátu?
Ako napísať vysvetlenie k žiadosti daňového inšpektorátu? KBK penále DPH pre právnické osoby
KBK penále DPH pre právnické osoby